El Extendido de Sangre Periférica
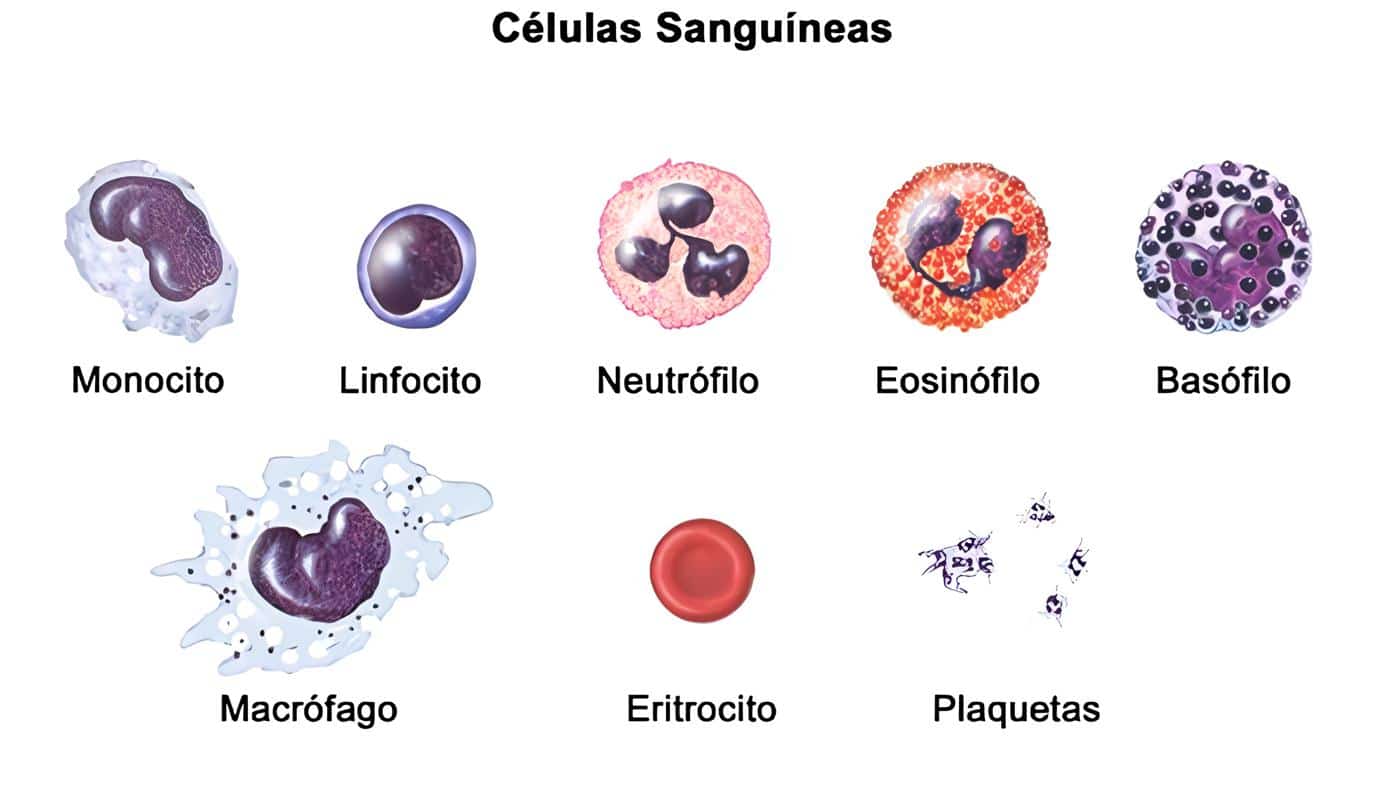
El ESTUDIO de las células sanguíneas por medio de la observación en un microscopio de las preparaciones coloreadas, conocido como ESP. Ha sido uno de los exámenes más antiguos e importantes en el laboratorio clínico. La práctica del ESP es de gran utilidad en hematología ya que permite visualizar la morfología celular. Estimar el número de células en sangre periférica y determinar la eventual presencia de elementos atípicos. Los resultados obtenidos con este estudio son un reflejo del funcionamiento de la médula ósea y de los factores que influyen en las diferentes poblaciones celulares (7, 8).
A pesar de su utilidad, bajo costo y fácil realización, el ESP está siendo desplazado actualmente por el uso de contadores electrónicos que emplean la impedancia eléctrica o la dispersión óptica para elaborar los hemogramas. Proporcionando un recuento celular muy preciso pero la información morfológica insuficiente.
Características microscópicas de las células sanguíneas
De esta forma, se pueden pasar por alto algunas características microscópicas de las células sanguíneas, que son de gran ayuda diagnóstica en diferentes entidades. Este método permite la evaluación del estado de maduración, las características tintoriales, el contenido de los gránulos, las inclusiones y FORMAS celulares. Todo lo cual se correlaciona con la fisiopatología de ciertas enfermedades, y puede proporcionar, como en el caso de las IDP (Tabla 1). Adecuada orientación en la evaluación de la respuesta inmune y un manejo inicial simplificado de los pacientes (5, 7, 9).
Tabla 1. Alteraciones en el extendido de sangre periférica observadas en las inmunodeficiencias primarias.
ALTERACIONES EN EL NÚMERO DE CÉLULAS |
DISMINUCIÓN |
Neutropenia |
|
Linfopenia |
|
||
Trombicitopenia |
|
||
AUMENTO |
Neutrofilia |
|
|
Linfocitosis |
|
||
Monocitosis |
|
||
Eosinofilia |
|
||
Trombocitosis |
|
||
TAÑAMO |
Aumento |
|
|
Disminución |
|
||
Inclusiones citoplasmáticas |
|
ID: Inmunodeficiencia.
Alteraciones en el Número de Células Sanguíneas en las IDP
Disminución en el número Neutropenia
Neutropenia, y su sinónimo granulocitopenia, indican una disminución significativa del recuento de Polimorfonucleares Neutrófilos (PMN) por microlitro de sangre periférica. La concentración de PMN en la sangre está influenciada por la edad, el sexo, la actividad física y los factores genéticos y ambientales. Se habla de neutropenia cuando el recuento de PMN es inferior a 1.5 x 103/mL y de “agranulocitosis” cuando está por debajo de 0.5 x 103/mL. Este último término habla de la ausencia absoluta de granulocitos, sin embargo, es usado en la práctica clínica para indicar una neutropenia severa (10-12).
La aparición de infecciones severas y recurrentes en el paciente neutropénico es una fuerte evidencia de la importancia de estas Células Sanguíneas en la defensa del hospedero. Los pacientes con recuentos de PMN inferiores a 500/mL tienen un riesgo severo de adquirir infecciones graves, y con menos de 200/mL se observa una respuesta inflamatoria prácticamente ausente. En general, el riesgo de infección de estos pacientes varía de acuerdo con la causa y la duración de la neutropenia, la cual se puede presentar por: alteraciones en la producción, destrucción excesiva en los tejidos periféricos, secuestro durante el proceso de maduración, distribución alterada y por combinación de estas anormalidades (6, 10, 12, 13).
Las neutropenias pueden ser de origen genético (IDP) o adquirido (medicamentos, infecciones, etc.). Dentro de las IDP que característicamente presentan neutropenia se incluyen las siguientes:
-
Enfermedad de Kostmann:
Es una entidad de herencia autosómica recesiva, originada por mutaciones en el gen que codifica para la elastasa de los PMN, ubicado en el cromosoma 19. Este defecto genera una alteración en la maduración de las células mieloides, traducido en una neutropenia severa o agranulocitosis. La granulocitopenia es severa y persistente, con recuentos de neutrófilos inferiores a 0.1 x 103/mL, y se puede acompañar de eosinofilia, monocitosis y anemia (12-14).
-
Síndrome de Shwachman:
Se hereda como un rasgo autosómico recesivo, asociado con mutaciones en el brazo largo de los cromosomas 6 ó 12. No obstante, el gen directamente afectado no se conoce todavía. Se caracteriza por una disminución en la producción y defectos en el crecimiento y diferenciación de los granulocitos, así como por insuficiencia pancreática y alteraciones osteocondrales. La neutropenia es constante, con recuentos de PMN menores de 1.0 x 103/mL. No se encuentra acompañada de monocitosis, pero se pueden observar anemia y trombocitopenia (1, 6, 12, 15).
-
Neutropenia Cíclica:
Esta enfermedad es también causada por mutaciones que afectan el gen de la elastasa del PMN. Es una FORMA particular de neutropenia, que varía de leve a severa, de comienzo temprano y que trascurre con oscilaciones periódicas de 21-30 días en el número de los PMN en sangre periférica. En muchos casos los reticulocitos, las plaquetas y otros leucocitos pueden presentar estos mismos ciclos de variación cuantitativa. Aunque el número de PMN es recurrentemente bajo, su función es normal. El diagnóstico sólo puede ser establecido después de realizar recuentos repetidos de los PMN en sangre periférica (tres por semana, durante seis semanas) en los pacientes con antecedentes familiares de la enfermedad o con un cuadro clínico sospechoso de ella (3, 6, 12, 13).
-
Neutropenia Familiar Benigna:
Es una enfermedad de carácter crónico y benigno, que se hereda como un carácter autosómico dominante. Generalmente, no se acompaña de síntomas y si se presentan son leves. Esta alteración se ha atribuido a un defecto en el desarrollo de los granulocitos en la médula ósea, pero el defecto genético específico no se conoce. El recuento total de leucocitos es normal, con una reducción marcada de los PMN y un incremento relativo de los monocitos y linfocitos. En el ESTUDIO de la médula ósea se observa una celularidad normal, con un desplazamiento a la izquierda de los precursores de los PMN (1, 3, 6).
-
Mielokatexis:
Desorden congénito, de carácter autosómico dominante, caracterizado por una apoptosis generalizada de los granulocitos debida a la expresión disminuida de la proteína bcl-X en las células precursoras de la médula ósea. Esta inmunodeficiencia presenta una neutropenia persistente desde la infancia. Los PMN de estos individuos muestran cambios degenerativos, tales como: permeabilidad aumentada a los colorantes, vacuolas citoplasmáticas, núcleo picnótico, hipersegmentado y cuyos lóbulos están conectados por filamentos delgados (16).
Existen además otras IDP en las cuales la presencia de neutropenia es característica de alguna etapa de la enfermedad:
Síndrome de Chediak-Higashi, agamaglobulinemia ligada al cromosoma X, hipoplasia cartílago-pelo, inmunodeficiencia común variable, síndrome de hiperinmunoglobulinemia M (Hiper-IgM) y deficiencia de adhesión leucocitaria tipo III (2, 13, 17).
Parte del éxito en el diagnóstico oportuno y el manejo apropiado de los pacientes neutropénicos reside en la notificación inmediata de los estados severos de neutropenia (considerados una emergencia médica) y en la identificación de la causa en el menor tiempo posible (12).