Paciente con Enfermedad de Cori-Forbes, Tipos de Glucogenosis
| Tipo: Nombre | Enzima afectada | Órgano primario | Manifestaciones |
| Tipo 0 | Glicógeno sintetasa | Hígado | Hipoglicemia, muerte temprana |
| Tipo la: Von Gierke´s | Glucosa 6 Fosfata | Hígado | Hepatomegalia, Falla renal, Disfunción del trombocito |
| Tipo Ib | Microsomal Glucosa 6 Fosfatasa Tranlocasa | Hígado | Similar a la, más neutropenina |
| Tipo Ic | Microsomal Transportados Pi | Hígado | Similar a la |
| Tipo II: Pompe´s | Lisosomal -1,4- Glicosidada Maltasa Ácida |
Música caridaco y esquelético | Forma Infantil=muerte; Forma Juvenil= miopía, Forma adulta= Adult form= distrofia muscular. |
| TIPO III: Coris´s or Forbe´s | Enzimas desramificadora | Hígado, Músculo esquelético y cardiaco | Hepatomegalia, Cirrosis. |
| TIPO IV: Anderson´s | Enzima Ramificadora | Hígado, Músculo | Hepatomegalia, Cirrosis. |
| TIPO V: Mc Ardle´s | Fosforilasa hepática | Hígado esquelético | Calambre y dolor inducidos por el ejercicio, Mioglobbinuria |
| Tipo VI: Her´s | Fosforilasa hepática | Hígado | Hepatomegalia |
| Tipo VII: Tarui´s | PFK-muscular | Músculos Glúobulos rojos | Similar a V, más anemia hemolítica |
| Tipo Vlb, VIII otipo IX | Fosforilasa kinasa | Hígado, Leucocitos Músculo | Similar a VI |
| x | PKA | HígadoMússculo | Hepatomegalia |
[column size=”1-2″ style=”0″ last=”0″]Glucogenosis Tipo III
Sinónimos:
Enfermedad de Cori-Forbes
Enfermedad de Forbes
Enfermedad de Cori
Deficiencia de amilo1,6 glucosidasa
Deficiencia de AGL
Deficiencia GDE
Deficiencia de la enzima desramificadora[/column]
[column size=”1-2″ style=”0″ last=”0″]Definición
Deficiencia de la enzima desramificante, amilo-alfa-1,6-glucosidasa.
El gen causante de la glucogenosis tipo III fue identificado y clonado en el cromosoma humano 1p21. Los subtipos de la enfermedad dependen de la mutación producida en este gen.[/column]

Organismo: Homosapiens.
Código genético: 1
Linage: Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Primates; Catarrhini; Hominidae; Homo.
Frecuencia
Incidencia en 2,3 niños por 100.000 nacimientos por año y en los Judíos no Ashkenazi, una frecuencia de 1 por 5.400 personas y en el Norte de África con una frecuencia de 1:53
División
IIIa Deficiencia de la enzima en todos los tejidos.
IIIb deficiencia de la enzima en el hígado.
Clínica
Tipo I (Forma Infantil)
Manifestación en los primeros cinco (5) meses de vida.
Hepatomegalia y/o esplenomegalia.
Hipoglicemia que se presenta principalmente en ayunas.
Elevación de los triglicéridos y el colesterol.
Hipotonia infantil con pobre control de los músculos de sostén de la cabeza, debilidad generalizada.
Pueden presentar cardiomegalia.
Retardo del desarrollo.
Tipo II (Forma Juvenil)
Hepatomegalia.
Puede presentar síntomas en la infancia que tienden a desaparecer en la pubertad.
Moderada a marcada.
Puede progresar a cirrosis o adenoma hepático.
Hipotonía
Mínima durante la infancia.
Caracterizada por progresiva debilidad muscular y emaciación.
Cardiomegalia:
Mínima durante la infancia.
En raras ocasiones conduce a la muerte súbita.
Es más común en adultos.
Otras:
Tendencia al sangrado.
Facies de querubín.
Abdomen globoso.
Baja estatura.
Infecciones recurrentes.
Laboratorio
Suero
Hipoglicemia, lactato normal, cetonas y hiperlípidemia.
Hiperbilirrubinemia conjungada.
Sin acidosis.
Electrocardiograma
Puede ser normal o presentar manifestaciones inespecíficas.
Rayos X de Tórax
Cardiomegalia.
Test de glucosa en sangre:
La medición de los niveles de glucosa 2 horas después de una comida – incremento de los niveles de glucosa.
La medición de los niveles de glucosa durante la noche – pueden estar planos (esto puede diferenciar esta glucogenosis de la tipo uno, debido a que en esta última, después de ambos procedimientos, los niveles de glucosa pueden estar planos).
Diagnóstico
Demostración
De la acumulación de glucógeno en el hígado y/o músculo
De la deficiencia de amilo-alfa-1,6-glucosidasa en hematíes
Presencia de oligosacáridos ricos en glucosa en la orina.
El diagnóstico enzimático se realiza mediante la demostración del mal funcionamiento de la enzima recolectada de la biopsia de hígado o músculo estriado de un paciente afectado, al compararlo con la actividad de la enzima de un paciente normal. El resultado se determina en dos semanas.
Biopsia Hepática
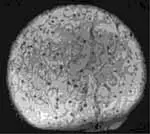
figura1. Biopsia del hígado que muestra citoplasma vacuolado de los hepatositos con núcleo central. (H&E’400)
Diagnósticos Diferenciales
Intolerancia a la glucosa
Deficiencia de la glucosa 6 fosfatasa
Deficiencia de la glucosa 6 fosfatodeshidrogenasa
Glucogenosis tipo II
Glucogenosis Tipo IV
Glucogenosis Tipo Ia
Glucogenosis Tipo Ib
Glucogenosis Tipo V
Glucogenosis Tipo VI
Glucogenosis Tipo VII
Hepatocarcinoma primario
Falla hepática
Hipoglicemia
Complicaciones
Carcinoma hepatocelular
Falla hepática
Tratamiento
El objetivo del tratamiento es mantener el equilibrio del metabolismo de los glúcidos y limitar la acidosis láctica y la cetosis sanguínea. La hipoglicemia y la acidosis láctica se evitan mediante una alimentación frecuente durante el día y algunas comidas nocturnas; solo en los casos más severos se utiliza alimentación continua por sonda nasogástrica en la noche.
Régimen con 50 – 60% de hidratos de carbono y dextrinomaltosas y no debe contener galactosa o fructuosa. El aporte de proteínas puede aumentarse relativamente de manera que en algunos casos llega a cubrir el 25% de las necesidades energéticas. Agregar al final de las comidas azúcares que se absorben lentamente, como la fécula de maíz no cocida, los tallarines o el couscous parcialmente cocidos permiten prolongar el período de normoglicemia postprandial.
Los niños con descompensación aguda con hipoglicemia y acidosis láctica deben ser hospitalizados y ser tratados con glucosa y bicarbonato de sodio por vía intavenosa. En los niños en los cuales el tratamiento dietario no es suficiente para evitar las hipoglicemias severas que terminan en estatus convulsivos o coma o en aquellos en los que se encuentra falla hepática o carcinoma hepatocelular el único tratamiento existente es el transplante hepático.
Pronóstico
En los estudios que se han llevado a cabo acerca de esta enfermedad se ha llegado a la conclusión de que es una de las enfermedades más benignas y controlable fácilmente si se recibe una dieta adecuada, a menos de que se presente una de sus complicaciones, en la cual el pronóstico cambia pudiendo llegar a ser una enfermedad rápidamente fatal.
Bibliografía
1. Aminoff MJ: Electromyography in Clinical Practice. New York, NY. Churchill Livingstone; 1998
2. Chen Y; Glycogen Storage Diseases In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill, Inc; 2001: 1521 – 1551.
3. Levin S, Moses S, Chayoth R: Glycogen storage disease in Israel. A clinical, biochemical and genetic study. Israel Journal of Medical Science 1967; 3 – 397.
4. Lucchiari S, Fogh I, Prelle A, Parini R, Bresolin N, Melis D, Fiori L, Scarlato G, Comi GP. Clinical and genetic variability of glycogen storage disease type IIIa: seven novel AGL gene mutations in the Mediterranean area. Am J Med Genet 2002 May 1; 109 (3): 183 – 190.
5. Horinishi A, Okubo M, Tang NL, Hui J, To KF, Mabuchi T, Okada T, Mabuchi H, Murase T. Mutatinal and haplotype analysis of AGL in patients with glycogen storage disease type III. J Hum Genet 2002; 47 (2): 55 – 59.
6. Descripción de los tipos de glucogenosis. www.ucip.net/aeeg/glucogen
7. Forbes Disease –glycogenosis III. www.icondata.com/health/pedbase/filos/FORBESDI
8. Doctor SOTELO CRUZ, Norberto. Lactante con hepatomegalia www.geocities.com/rgarayc/may01l
9. Alpa S. Amin, Kasturi L., Kilkarni A. V., Ajmera N. K. Glycogen storage disease type III. www.indianpediatrics.net/june2000/june-670-673
10. Amylo –1,6 –glucosidase, 4 –alpha –glucanotransferase isoform 3; glycogen debraching enzyme; amylo – 1,6 –glucosidase, 4 –alpha –glucanotransferase (glycogen debraching enzyme); amylo –1,6 –glucosidase, 4 –alpha –glucanotransferase (glycogen debraching enzyme, glycogen storage disease type III). www.ncbi.nlm.nih.gov/cgi-bin/Entrez/framik?gi=4557285&db=Protein
11. 2322400 glycogen storage disease III. www.ncbi.nih.gov/htbin-post/0mim/dispmim?2324000#TEXT
12. María Sepúlveda, Nora Yepes, Fernando Gutiérrez. Hepatopatía crónica en niños. Justificación para un programa de transplante hepático infantil. https://medicina.udea.edu.co/iatria/Marzo2002/IATREIA%20Vol%2015%20N°1%20Art.4.pdf-
13. Glycogen storage disease, type III (GSD III) www.duke.edu/~mdfeezor/dukemedicalgenetics/gsdiii
14. P. Jara. Hepatopatía pediatricas. www.aeeh.org/trat_enf_hepaticas/c-53.pdf
15. Instituto de bioquímica clínica. Servicio de diagnóstico de enfermedades metabólicas hereditarias. www.sen.es/recursos/bioq/bioquimical
6. Castellanos A, Ortiz F, García M, Prieto F, Santidrián J, Mazorra F. Evaluación de la autopsia en la unidad de cuidados intensivos pediátricos. www.aeped.es/anales96-99/suma/vol46/46-3/46-3-4.pdf
17. Type III glycogen storage disease (GSD type III). www.agsdus.org/body_typeii_12
18. Glycogen storage disease, tipe III. www.emedicine.com/med/topic909
19. What is glycogen storage disease (GSD)? www.agsdus.org/body_whatis_1l
20. Type IIIb glycogen storage disease associated with end – storage cirrhosis and hepatocellular carcinoma. www.geocities.com/HotSprings/Spa/7563/meta06l
21. Glycogen storage disease. www.ahealthyme.com/topic/topic100586878
CLIC AQUÍ Y DÉJANOS TU COMENTARIO