Guía de Manejo de Errores Congénitos del Metabolismo, Diagnóstico
Los hallazgos clínicos suelen ser inespecíficos y parecidos a infecciones generalizadas en lactantes. Los RN suelen ser normales al nacer, sin embargo unas horas después del alumbramiento, pueden presentar letargo, rechazo al alimento, convulsiones y vómitos. La historia de un RN previamente sano con deterioro de su estado, debe hacer sospechar un ECM5.(figuras No 3-4 y 5).
Muchos de los ECM, incluyendo defectos del ciclo de la úrea, acidemias orgánicas y ciertos desórdenes del metabolismo de los aminoácidos, se presentan en niños pequeños con síntomas de encefalopatía metabólica aguda y crónica; otros síntomas típicos incluyen letargo, inapetencia, apnea, taquicardia y vómitos recurrentes. Acidosis metabólica y/o hiperamonemia es observada a menudo en estas condiciones pero hay excepciones, como la hiperglicinemia no cetósica y déficit de cofactor molibdeno. La hipoglicemia puede ser el hallazgo predominante en algunas ECM como enfermedad de depósito del glucógeno, defecto en la gluconeogénesis, y defecto de oxidación de ácidos grasos. Algunos desórdenes tienen manifestaciones tardías con variabilidad clínica, como muerte súbita, episodio semejante al Síndrome de Reye o cardiomiopatía. La galactosemia, tirosinemia hereditaria o hemocromatosis neonatal se presentan con ictericia u otra manifestación de disfunción hepática. Un subgrupo de desórdenes de depósito lisosomal se manifiestan tempranamente con organomegalia e hidrops. Patrones específicos de dismorfología se presentan en el Síndrome de Zellweger y Síndrome de Smith-Lemli-Optiz4.
La consanguinidad, antecedentes de muertes neonatales en hermanos o varones afectados del lado materno deben hacer sospechar ECM. Otros familiares con RM, deficiencia neurológica o preferencia dietética poco habitual pueden expresar una manifestación sutil de la misma patología. Aunque la mayoría de neonatos con ECM tienen antecedente familiar negativo2
Manifestaciones clínicas en ECM:
1. Encefalopatía metabólica aguda: Es la manifestación más importante de las acidemias orgánicas, defecto del ciclo de la úrea y ciertos desórdenes del metabolismo de los AA, presentándose con signos que amenazan la vida por efecto tóxico de metabolitos acumulados en SNC, que en la vida neonatal atraviesan la placenta y son metabolizados por la madre, la cual hace ver al RN normal cuando nace. El inicio de los síntomas va desde horas a meses.
Los hallazgos iniciales son letargo, inapetencia, debilidad y síntomas que semejan sepsis, progresando hasta coma. Otros síntomas de disfunción del SNC son convulsiones, trastornos del tono muscular, edema cerebral y en algunos hemorragia intracerebral que ocurre infrecuentemente. Síntomas respiratorios como apnea central y disnea por encefalopatía metabólica, la taquipnea ocurren por acidosis que puede llevar a alcalosis respiratoria4. El vómito ocurre menos frecuente en RN que en niños mayores, hace parte del diagnóstico diferencial como estenosis pilórica e intolerancia a la fórmula láctea4.
2. Olor anormal: El cual es percibido por las madres o enfermeras, se presenta en cuadros agudos en acidemia isovalérica, acidemia glutárica tipo II y enfermedad de orina en jarabe de arce4.
Fenilcetonuria olor a moho.•Jarabe de arce olor a caramelo quemado.
Hipermetionemia olor a repollo descompuesto.
Acidemia butírica o isovalérica olor a pies sudados o queso rancio
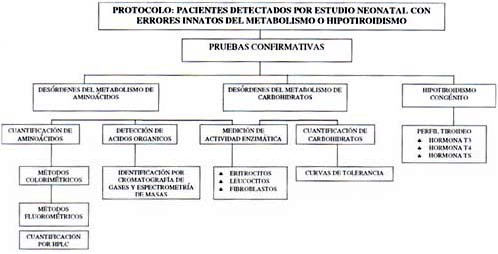
Figura No.3
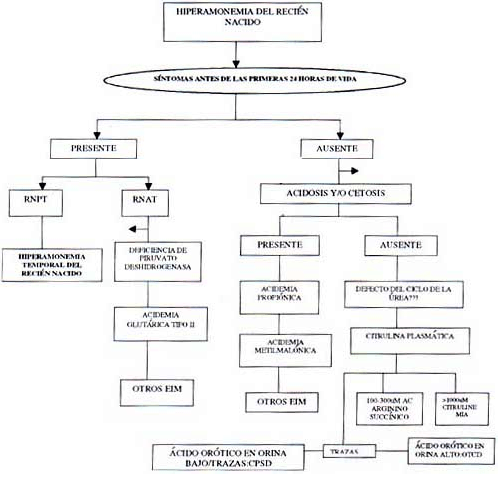
Figura No. 4
3. Dismorfología:
El Síndrome de Zellweger y la adrenoleucodistrofia, las cuales producen hipotonía y epicanto, manchas de brushfield, fontanela amplia, línea simiana y quistes renales4.
Acidemia glutárica y aciduria 3 OH isobutírica: Semejan el SS de alcohol fetal4.Acidemia glutárica tipo II. Frente amplia, hipertelorismo, cejas de implantación baja, megalonefronia, hipospadias y pies en mecedora4.
SS de Smith-lemi-optiz: fascies dismórficas, paladar hendido, cardiopatía, hipospadia, polidactilia y sindactilia4.
Hiperglicinemia no cetósica: Agenesia del cuerpo calloso4.
Paraclínicos: Muestras a obtener de todo niño con sospecha de error congénito del metabolismo y se deben realizar entre 72 h y 1 mes2,4.
1. Amonio: La hiperamonemia siempre se encuentra en paciente con letargo, vómito u otra evidencia de encefalopatía. Las causas más frecuentes de hiperamonemia en ECM son los defectos del ciclo de la Úrea y las acidemias orgánicas. El tiempo en que se inician los síntomas son una ayuda diagnóstica, en las primeras 24 horas indicarían: la acidemia glutárica tipo II, déficit de la piruvato carboxilasa o la hiperamonemia transitoria neonatal en RNPT. La medición de AA es útil para diferenciar los defectos del ciclo de la Úrea4.
2. Gases Arteriales: La acidosis metabólica es el segundo hallazgo de laboratorio de importancia en ECM durante el episodio agudo de enfermedad acompañado de anión gap elevado (El dx diferencial lo hacen la EDA y la ATR). Los ECM son acidosis metabólica y lactato elevado son las acidemias orgánicas (acidemia metilmalónica, propiónica e isoválerica) o defectos en la oxidación de ácidos orgánicos.
Defectos en el metabolismo del piruvato o en la cadena respiratoria llevan a acidosis láctica, la diferenciación entre los desórdenes de este grupo se pueden realizar midiendo el piruvato en plasma y calculando la relación lactato/piruvato, una relación 25 sugiere deficiencia de la piruvato carboxilasa o de la cadena respiratoria.
No todos los ECM se acompañan de hiperamonemia o acidosis como es la hiperglicinemia no cetósica4.
3. Glicemia: hipoglicemia, aunque se asocia a desórdenes del metabolismo de las proteínas, es más frecuente en desórdenes de carbohidratos o oxidación de ácidos grasos, los más frecuentes y conocidos son las enfermedades de depósito del glucógeno; la hipoglicemia es debida a la imposibilidad para formar glucosa a partir de glucógeno y es más severa en períodos de ayuno. La hipoglicemia se puede encontrar en galactosemia o intolerancia a la fructosa, aunque los síntomas ocurren posterior a la introducción de la fructosa (sucrosa) y galactosa en la dieta4.
4.Otros: Cuadro hemático, parcial de orina, electrolitos séricos, sustancias reductoras en orina, cetonas en orina, cuantificar aminoácidos en orina y sangre, ácidos orgánicos en orina, lactato plasmático4.
5. Hallazgos de RNM:
1. Alteración de la sustancia gris profunda, sustancia blanca, compromiso globus pallidus: Acidemia metilmalónica y propiónica, enfermedad de orina en jarabe de arce23.
2. Alteración de la sustancia blanca periférica, con perímetro cefálico normal: Galactosemia23.
3. Alteración de la sustancia blanca profunda, con tálamo normal y compromiso no específico del tallo: Fenilcetonuria, enfermedad de orina en jarabe de arce compromiso de cerebelo y pedúnculos 3. Alteración de la sustancia blanca profunda, con tálamo normal y compromiso no específico del tallo: Fenilcetonuria, enfermedad de orina en jarabe de arce compromiso de cerebelo y pedúnculos 3. Alteración de la sustancia blanca profunda, con tálamo normal y compromiso no específico del tallo: Fenilcetonuria, enfermedad de orina en jarabe de arce compromiso de cerebelo y pedúnculos23.
4. Alteración de la sustancia blanca, con patrón inespecífico difuso unilateral, asimétrico: Hiperglicinemia no cetósica y desórdenes del ciclo de la úrea23.
CLIC AQUÍ Y DÉJANOS TU COMENTARIO