Inmunoalergia. 09 nº 2
Lo más reciente

Revista de Inmunología: Comité Editorial, Vol. 09 No. 2
Volumen 9. Número 2, Junio 2000 JUNTA DIRECTIVA 2000 – 2002 PRESIDENTE Eduardo Egea VICEPRESIDENTE Ricardo Cardona SECRETARIA EJECUTIVA Mario Sánchez...
Editorial: III Congreso de la Asociación Colombiana de Alergia, Asma e Inmunología
Se acaba de realizar el III Congreso de la Asociación Colombiana de Alergia, Asma e Inmunología en la ciudad de...

Proteínas Alergénicas de Fraxinus Sinensis y Cecropia Sy y Reactividad Cruzada entre Oleáceas
Adriana Rodríguez Ciódaro MScD1 Francisco J. Leal Quevedo MD2 En este trabajo se determinó la capacidad alergénica del polen de...

Obtención de los granos de polen
Métodos El polen de F. sinensis fue recolectado tomando flores masculinas de diferentes árboles, suspendiéndolas y manteniéndolas a temperaturas elevadas...

Inhibición del RAST
Para determinar la reacción cruzada se utilizaron extractos de F. sinensis (preparado como se describe anteriormente), velutina, pennsylvanica, latifolia, americana...
Inmunodetección en el Fraxinus Sinensis y Cecropia Sy
Los seis pacientes positivos a Fraxinus mostraron reconocimiento por IgE específica de bandas de proteínas en un rango de peso...
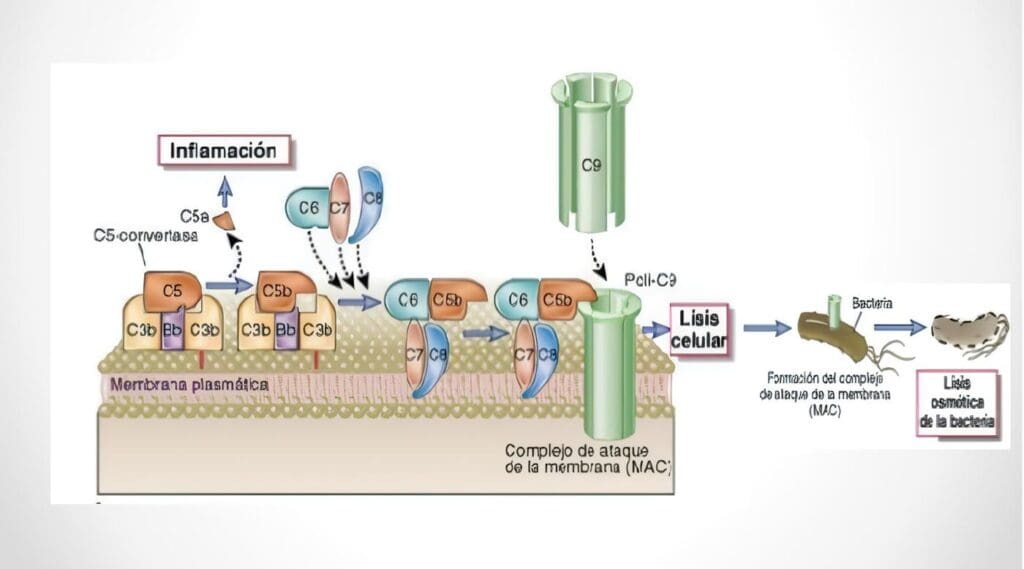
Guía de Estudio y Manejo del Paciente Sospechoso de Presentar Alteraciones en el Sistema del Complemento
Complejo de Ataque a Membrana Helí Salgado, Carlos J. Montoya, Juan A. López, Pablo J. Patiño Grupo de inmunodeficiencias primarias,...
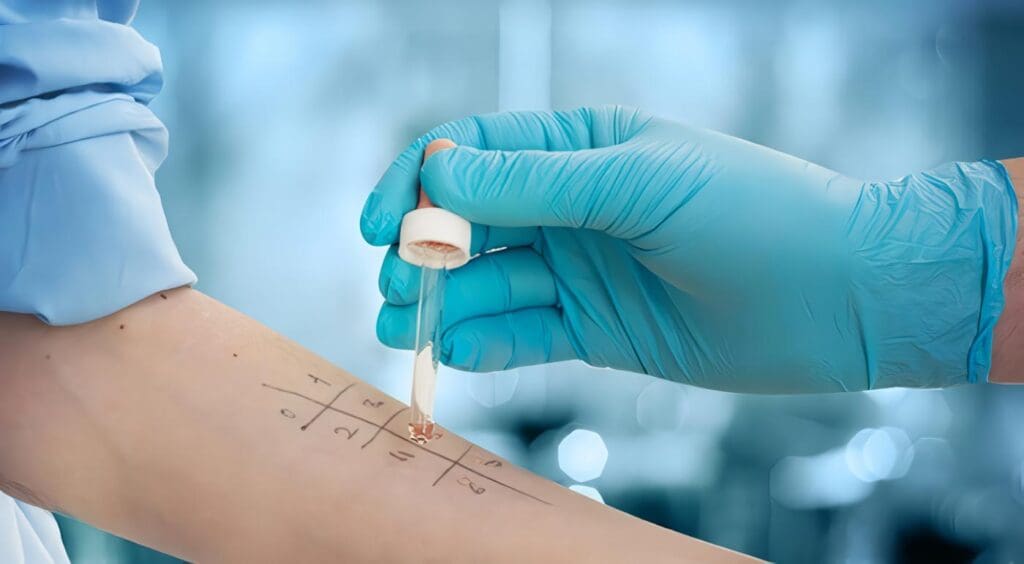
Utilidades de las Pruebas In Vivo en Alergia a Betalactámicos
Joaquín Sastre D. MD. 1 Ricardo Cardona V. MD. 2 1 Fundación Jiménez Díaz. Madrid – España 2 Unidad de...

Alergia a Betalactámidos, Prueba In Vitro
La determinación de IgE específica a penicilina G y V, con distintos conjugados como albúmina o polilisina, tiene una menor...
Que tan Lejos se ha LLegado en el Conocimiento de las Inmunodeficiencias Primarias
Diana García de Olarte Pediatra, Doctor en Ciencias Básicas Biomédicaz Profesor Facultad de Medicina, Universidad de Antioquia Hace cinco décadas,...