Gabriel Toro González*
Arturo Díaz Gómez**
Cecilia Saad Acosta***
(Reproducido con autorización de Acta Neurológica Colombiana Vol. 18 No. 4, 2002).
Introducción
La creencia mejor respaldada actualmente nos indica que existe un grupo de enfermedades causadas por anormalidades en el procesamiento de las proteínas.
En la década 1950-1960 D. Carleton Gajdusek y Vincent Zigas comenzaron a estudiar en Nueva Guinea una enfermedad fatal del sistema nervioso que se conoce con el nombre de Kuru que estaba diezmando a la tribu Fore, comunidad de unos 12.000 nativos (Figura 1).
Gajdusek y Zigas observaron que el Kuru no correspondía a ningún modelo genético conocido y con la colaboración del antropólogo R. Glasse pensaron que el canibalismo ritual practicado por los Fore podría ser la causa de la transmisión de esta enfermedad, lo cual resultó cierto porque el Kuru prácticamente desapareció tras tres décadas de abolidos estos hábitos. (Lea también: La Teoría del Prión, Insomnio Fatal (IF))
Desde el primer momento se notó que el Kuru tenía características comunes con el Scrapie y que había semejanza entre estas dos entidades y la enfermedad de Creutzfeldt-Jakob (ECJ), una demencia de progresión rápida, no limitada geográficamente, descrita a principios del siglo XX, las tres aparentemente causadas por infecciones virales lentas con largos períodos de incubación medibles en años y un avance irreversible de los síntomas.
Este pensamiento, por supuesto, se apoyaba en la afirmación formulada en 1939 por Mc Farlane Burnet de un posible mecanismo de acción lenta o temperada, inicialmente ejemplificada en los virus Herpes y en la propuesta que unos 15 años más tarde hiciera el veterinario islandés B. Sigurdsson de llamar Enfermedades Virales Lentas a las entidades que exhibieran tal comportamiento.
Philip Duffy y colaboradores en la Universidad de Columbia, en 1974 comunican la primera observación de transmisión de la demencia de Creutzfeldt –Jakob de humano a humano a través de un injerto de córnea y a esta se sumaron otras posibilidades de transmitir el agente causal, entre ellas electrodos contaminados, hormona de crecimiento, transplante de duramadre.
Pese a esta demostración de transmisibilidad la teoría viral desde el comienzo encontró fuerte oposición; lo cual no impidió que la comunidad científica comprendiera el alcance de esta ejemplarizante investigación y concediera por ella a DC Gajdusek el Premio Nobel de Medicina en 1976.
El punto de partida de este monumental trabajo fue entregado a la comunidad científica desde 1981 en el libro cuya carátula vemos en la Figura 2. En 1970 J.S. Griffith y R. Latarget plantean que estos agentes infecciosos carentes de ácidos nucléicos en vez de estructura viral podrían tener naturaleza esencialmente protéica

Figura 1. Aldea del grupo lingüístico Fore en la Nueva Guinea una de las áreas donde el Kuru probó en el pasado su capacidad devastadora (cortesía de D.C. Gajdusek).
El punto de partida de lo que podríamos llamar el 2º capítulo de esta problemática o si se prefiere el presente de la misma, nos lo cuenta el propio “creador de los Priones” así: “Yo me interesé por primera vez en las enfermedades por priones en 1972, cuando siendo residente de Neurología en la Universidad de California vi morir a uno de mis pacientes debido a la enfermedad de Creutzfeldt-Jakob” (Stanley B. Prusiner-Premio Nobel de Medicina 1997).

Figura 2. Carátula del libro que condensa la historia de los primeros años de investigación en Kuru (editado en 1981). Este ejemplo de búsqueda incansable es el punto de partida del estudio del grupo de entidades que durante tres décadas se denominaron Enfermedades virales lentas, la mayoría de ellas hoy consideradas priónicas..
En 1982, S.B. Prusiner descubrió y aisló partículas protéicas infecciosas y les asignó el nombre de “Priones”.
Prusiner sugirió que una variedad de enfermedades neurodegenerativas con componente genético, infeccioso o esporádico podría ser el resultado de dicha infección, involucrando procesos anormales de proteínas neuronales.
En el lapso 1982-2002 su densa contribución a este problema ya guarda paralelo con la aportada por D.C. Gajdusek y su grupo en las tres décadas anteriores.
Las enfermedades humanas causadas por priones incluyen Kuru, Creutzfeldt-Jakob esporádico (CJe) y familiar (CJf), Síndrome de Gertsmann – Straussler – Scheinker (GSS), insomnio fatal (IF) y una nueva variante de Creutzfeldt–Jakob (vCJ) descrita recientemente (cuadro 1), constituyen un grupo de desórdenes con perfil de Encefalopatías Subagudas Enpongiformes Transmisibles (ESET) de evolución lenta e irreversible que conducen a la muerte y que suelen afectar otros vertebrados.
En 1936 se demostró transmisión de Scrapie de oveja a oveja y en 1960 Cuillé y Chelle transmitieron Scrapie (tembladera del cordero) a cabras sanas por medio de la inoculación intraocular de medula espinal de oveja infectada.
La Encefalopatía Espongiforme Bovina (EEB) aparecida por primera vez en 1985 en el Reino Unido es desde el punto de vista clínico y por su patología idéntica al Scrapie pero en bovinos, cuyo comportamiento “pseudoneurótico” le ha valido el calificativo de “vacas locas”.

Cuadro 1. Enfermedades humanas de etiología priónica y su patogenia.
Este comportamiento del animal atáxico y tembloroso incluye excitabilidad, agresividad y finalmente imposibilidad para sostenerse en pie y para marchar.
Muy significativos hechos en varios zoológicos ingleses precedieron la identificación de la EEB, el primero de ellos fue la muerte de un nyala ungulado salvaje africano en Marwell cerca de Winchester, luego fallecen 3 bisontes americanos en el zoológico de Londres, a los cuales se suman una serie de antílopes, alces y oryx; la autopsia de estos animales comprobó una encefalopatía espongiforme.
Todos ellos estaban recibiendo desde 1981 alimentos que incluían proteínas provenientes de ovejas afectadas de scrapie; por idéntica razón se ha considerado que la enfermedad ha pasado de la oveja al bovino. El cuadro 2 consigna las que hasta el momento se consideran enfermedades de origen priónico en animales.
Priones
Es bueno anticipar que las enfermedades que hoy se atribuyen a estos agentes han recibido sucesivas diferentes denominaciones, siempre basadas en su substrato neuropatológico y en la etiología que se les ha atribuido en diferentes etapas durante la segunda mitad del siglo XX (1950-2000).
Esta sinonimia hasta ahora es así:
1. Encefalopatías espongiformes,
2. Enfermedades virales lentas,
3. Demencias transmisibles,
4. Amiloidosis viral del sistema nervioso,
5. Enfermedades priónicas.
Los priones son el único ejemplo conocido de patógenos infecciosos que están desprovistos de ácidos nucléicos en contradicción con el dogma central de la biología el cual afirma que “todas las formas de vida desde los virus hasta las plantas y los animales superiores transmiten sus caracteres a las siguientes generaciones a través del DNA (excepcionalmente RNA)”.
Todos los otros agentes poseen genomas compuestos por RNA o DNA que dirigen la síntesis de su progenie. Siendo así, se comprende la polarización con la que la Teoría Prion está siendo recibida por la comunidad científica, veamos: “La evidencia hacia la hipótesis prion está obteniendo más y más solidez” (Charles Weissmann).

Cuadro 2. Enfermedades causadas por priones en animales y su patogenia.
“Algunos aspectos de la hipótesis prion están cayendo en el seno del pensamiento medioeval” (Laura Manuelides).
El agente priónico transmisible de las ESET es no convencional, entiéndase no ceñido al conocimiento biológico actual, teniendo en cuenta que carece de ADN y ARN, y por ello es resistente a la inactivación por procedimientos que modifican los ácidos nucléicos pero en cambio sensibles a los métodos disponibles para degradar proteínas, tiene largos periodos de incubación, no produce respuesta inmunológica ni inflamatoria, no tiene estructuras visibles al microscopio electrónico y presenta extrema resistencia a la radiación ultravioleta, rayos X y al formaldehído; esta resistencia física y química de este agente infeccioso tan inusual ha contribuido a que se desarrollen casos iatrogénicos.
Es justo anotar que estas calidades del agente infeccioso, aparte de su denominación Virus o Prion fueron tempranamente identificadas por DC Gajdusek y sus colaboradores.
Proteínas Priónicas
Los estudios de las ESET condujeron al descubrimiento de una proteína de 27 a 30 kD a la que se denominó proteína priónica [PrP]. La forma normal de esta proteína se encuentra codificada por un gen ubicado en el cromosoma 20.
El producto logrado es una proteína celular [PrPc] proteasa sensible acumulada especialmente en la membrana neuronal y en las sinapsis que tiene entre 33 y 35 kD. Es posible distinguir entonces dos formas de la PrP, una presente en organismos sanos y otra presente en organismos afectados, esta última se asimila a la llamada proteína priónica scrapie [PrPsc] o proteína priónica proteinasa K resistente, insoluble en detergentes no denaturantes.
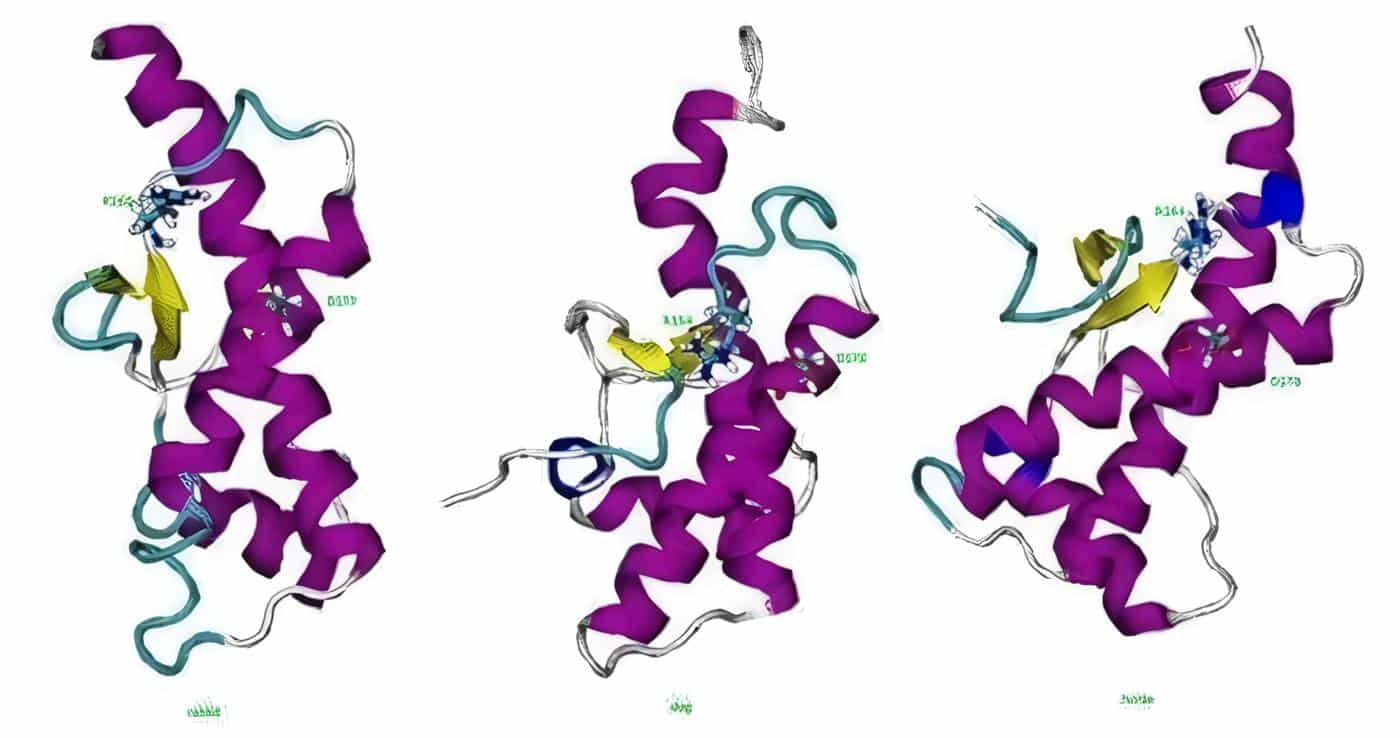
De la PrPc ya hay fragmentos incluidos en el PDB (Protein Data Bank) y en este momento se investigan fármacos que pudieran impedir la conversión de [PrPc] en [PrPsc]. La proteína priónica celular [PrPc] es una proteína proteasa sensible constituida por una sola cadena peptídica; presenta una estructura compacta con 4 hélices alfa (H1 a H4) anclada a la superficie de las neuronas por la glicoproteína I.
Consta de 253 aminoácidos en el hombre y es constante en diversas especies incluyendo humanos, ovejas, ratones, hamster y bovinos.
En los tejidos de los pacientes con ESET hay una conformación anormal parecida al scrapie designada como proteína priónica [PrPsc[, cuya concentración se incrementa progresivamente con la evolución de la enfermedad llegando a acumularse en forma de placas amiloides extracelulares después de polimerisarse.
Cabe agregar que investigadores que trabajan en fisiología de proteínas con plegamiento alterado han encontrado priones en levaduras y otros hongos sin causar lesión a este hospedero aparentemente.
Conversión de [PrPc] en [PrPsc]
En primer lugar se tiene claro que las moléculas mutantes no provienen de la transcripción génica tan pronto es sintetizada porque se esperaría que las personas que poseen mutación génica desarrollaran la enfermedad en la infancia; se sospecha que la mutación de la proteína resulta de la susceptibilidad de plegarse de alfa hélice a la hoja beta plegada.
Según la teoría de Prusiner de la sustitución de un aminoácido por otro en los bordes de las hélices la estructura protéica se desestabiliza aumentando la posibilidad de que se cambie su conformación plegándose sobre si misma según indican algunos ejemplos en el cuadro 1.
Para justificar la conversión de [PrPc] en [PrPsc] se han propuesto diversas hipótesis. Según la teoría pura del prión de Prusiner habría una interacción directa entre una molécula de [PrPc] y una de [PrPsc]; la [PrPsc] induciría la conversión de [PrPc] en una segunda molécula de [PrPsc], copia idéntica de la primera, así [PrPsc]+[PrPc]= nueva [PrPsc](efecto dominó multiplicador).
También se ha propuesto que la modificación estructural podría ser el resultado de un proceso de polimerización en cadena, iniciado por la [PrPsc] inoculada que actuaría como cristal iniciador.
Otros Investigadores apoyan la intervención de proteínas chaperonas, las cuales modificarían el plegamiento de [PrPc] o de su precursor; la [PrPsc] incluso podría ser la chaperona de [PrPc]. Queda claro que el punto fundamental para resolver sería impedir que la [PrPc] haga su transformación a [PrPsc], logro que aun parece lejano.
Pero el debate no se limita a lo expresado hasta aquí. Si los creadores y defensores de la Teoría Viral Lenta tuvieron que soportar tanta argumentación en contra y durante 30 años tantas preguntas quedaron sin respuesta, la situación no es del todo diferente ahora.
La máxima inconformidad ha sido suscitada por la Shattuck Lecture de mayo 17/2001, en la cual S. Prusiner sugiere que los Priones podrían explicar el origen de numerosas entidades cuya etiología guarda todavía muchos secretos.
Chiesa y Harris han subido la temperatura a esta controversia con afirmaciones como estas:
1. Se ha comprobado la presencia de [PrPsc] sin síntomas clínicos ni substrato neuropatológico de ESET,
2. Se ha observado ocasionalmente el cuadro clínico y el complejo neuropatológico de ESET con [PrPsc] escasa o indetectable en ratones inoculados con Scrapie y con EEB,
3. Ratones inoculados con priones de hamster adquieren niveles altos de [PrPsc], degeneración esponjosa y placas amiloides en cerebro pero no expresan síntomas clínicos,
4. En enfermedades priónicas familiares parece no ser necesaria la presencia de [PrPsc] para desarrollar ESET. Podrá PrPc inducir neurodegeneración sin tener que convertirse en [PrPsc]?
Algunos biólogos moleculares se preguntan hoy si la alteración en el plegamiento de proteínas puede hacer parte de un proceso patológico pero también fisiológico o normal.
Epidemiología
En Colombia no hay datos estadísticos acerca de las Encefalopatías Subagudas Espongiformes Transmisibles pero se han identificado algunos casos de enfermedad de Creutzfeldt-Jakob esporádica y hace 21 años el diagnóstico oportuno de Scrapie en un macho reproductor Cheviot importado de Escocia y las medidas adecuadas que se tomaron evitaron su diseminación.
En el año 2000 se han informado cerca de 400 casos de enfermedades priónicas en los Estados Unidos con una incidencia de menos de un caso por cada 100.000 habitantes.
La incidencia de ECJ esporádica es aproximadamente de 1 caso por cada millón de habitantes, pero en personas mayores de 60 años de edad alcanza 5 casos por millón; esta forma es lentamente progresiva causando la muerte usualmente en el primer año después del inicio.
La forma hereditaria, que aun genera controversia, comienza a edades más tempranas y tiene un curso más insidioso. El universo del problema causado por la Encefalopatía Espongiforme Bovina podría sintetizarse indicando que entre 1986 y 1998 causó la muerte de 170.000 animales en 34.000 hatos del Reino Unido y el sacrificio de 300-400 vacunos semanalmente.
Scrapie
Esta entidad apareció en 1732 en Inglaterra y Escocia y fue descrita por Teissier en 1810. Se ha detectado en Irlanda, Kenia, India, Sur Africa, Emiratos Arabes, Australia, Nueva Zelandia, en varios países de Europa Occidental. En Estados Unidos se presentó inicialmente en Míchigan en 1942, luego en California donde causó en 1954 la muerte de 576 animales.
Afecta ovinos, ocasionalmente caprinos causándoles excitabilidad, incoordinación para la marcha, dificultad para mantenerse en pie, temblor de la cabeza, ataxia especialmente de los miembros anteriores y muy marcado prurito cutáneo. La transmisión de oveja a oveja se comprobó en 1936 y de oveja a ratón en 1961.
La vacuolización neuronal encéfaloespinal es un aspecto muy relevante en su patología; este y los otros cambios patológicos fueron descritos inicialmente por Besnoit y Morel en 1898. No existe prueba de que el scrapie se haya transmitido al hombre (oveja-hombre). También se sabe que el ratón transgénico privado del gen de la PrP (ratón Knokout) no desarrolla enfermedad aunque se inocule con scrapie [PrPsc].